创新团队发现一类新型SHP2 tunnel位点变构抑制剂
2023年3月23日,杭州医学院药学院梁广团队陈凌峰课题组在药物化学权威期刊European Journal of medicinal chemistry(中科院一区TOP,IF = 7.088)在线发表题为“Discovery of a potent and selective allosteric inhibitor targeting the SHP2 tunnel site for RTK-driven cancer treatment”的研究论文,发现了一个对RTK驱动的疾病具有潜在开发价值的新型高选择性SHP2变构抑制剂。课题组研究生罗瑞祥、邵静静和马琳为共同第一作者。杭州医学院梁广研究员、陈凌峰副研究员以及浙江大学城市学院李杰教授为共同通讯作者。论文链接:https://authors.elsevier.com/sd/article/S0223-5234(23)00271-4。
SHP2(Src homology-2 domain-containing protein tyrosine phosphatase-2)是一种非受体蛋白酪氨酸磷酸酶。SHP2作为第一个报道的致癌酪氨酸磷酸酶,是人体内几乎所有RTK信号通路下游的关键蛋白,当SHP2的SH2结构域与RTKs和衔接蛋白上的pTyr相互作用时,SHP2被激活以去磷酸化其底物,进一步激活RAS-RAF-ERK途径促进癌细胞增殖。此外,SHP2缺失显著地抑制RTK驱动的癌细胞增殖,证明SHP2在RTK驱动的癌细胞增殖中发挥的重要作用。因此,开发特异性的SHP2抑制剂,用于治疗RTK驱动的疾病,具有重要的临床意义。
在已报道的代表性SHP2变构抑制剂(SHP099等)研发过程中,均需要对10万个以上的化合物库进行多轮高通量筛选,化合物的实际变构位点也需要通过X射线晶体衍射确定,因而需要消耗大量人力物力。课题组通过直接针对SHP2的tunnel变构位点,开展了虚拟筛选工作,得到了对SHP2具有变构抑制能力的先导化合物70。大大节省了高通量筛选所需要的时间。
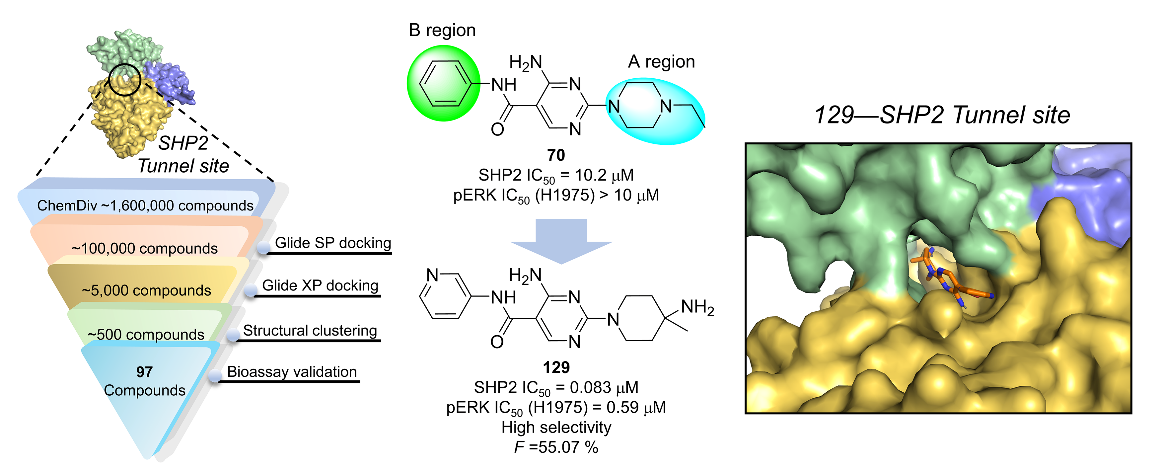
针对Hit compound 70两个位点的结构优化,最终获得一个高选择性、高生物利用度的新型SHP2变构抑制剂129。后续对PTPN12、ACP1等其他蛋白酪氨酸磷酸酶以及97种激酶谱的筛选发现候选化合物129对SHP2有高度的选择性。并且体内口服生物利用度达到55.7%。该类化合物目前已经申请结构专利保护(申请号: 202310275813.0)。后续课题组将评价化合物129在SHP2相关的炎症性疾病中的治疗效果。