Adv Sci 杭州医学院梁广/杭州师范大学王怡团队揭示蛋白激酶调控动脉粥样硬化的新机制
动脉粥样硬化(Atherosclerosis)是世界范围内发病率和死亡率较高的心血管疾病。动脉粥样硬化病变进程中的一个决定性事件是慢性的炎症反应,其主要特征是炎症细胞(包括巨噬细胞、中性粒细胞与单核细胞)的浸润和促炎因子的分泌。尽管动脉粥样硬化已有治疗方法,但病情的复发和再住院率依然居高不下,迫切需要新的治疗靶点和策略。蛋白激酶(Protein kinases, PKs)在动脉粥样硬化的病理过程中起着至关重要的推动作用,有望从中找到防治新靶点和新策略。
2025年4月9日,杭州医学院梁广教授联合杭州师范大学王怡教授研究团队在Advanced Science杂志在线发表了题为“Macrophage WEE1 Directly Binds to and Phosphorylates NF-κB p65 Subunit to Induce Inflammatory Response and Drive Atherosclerosis”的研究论文。该研究报道了巨噬细胞中的蛋白激酶WEE1在动脉粥样硬化中的作用,发现巨噬细胞特异性缺失或抑制WEE1激酶活性的药理作用可以通过抑制NF-κB介导的炎症来减轻动脉粥样硬化的进展。机制上,激活后的WEE1能够直接结合p65并特异性磷酸化其S536位点,随后激活NF-κB及下游的炎症因子风暴。这一突破性发现不仅阐明了WEE1- p65信号轴在动脉粥样硬化中的分子调控机制,更为动脉粥样硬化的防治提供了新的理论依据和潜在治疗靶点,具有重要的临床转化价值。

研究者基于多个动脉粥样硬化小鼠模型的RNA测序数据,通过激酶富集分析(KEA)筛选并鉴定了蛋白激酶WEE1的S642位点磷酸化水平在人和小鼠的动脉粥样硬化斑块巨噬细胞中显著上调。利用巨噬细胞特异性WEE1敲除小鼠原代细胞并通过高通量RNA测序分析,研究者发现WEE1可能调控巨噬细胞炎症反应。进一步的实验证实,WEE1促进了oxLDL诱导的巨噬细胞炎症反应,并且WEE1的这一作用依赖于其S642位点磷酸化后产生的激酶活性。而WEE1的特异性抑制剂MK1775可缓解oxLDL诱导的巨噬细胞炎症反应(图1)。
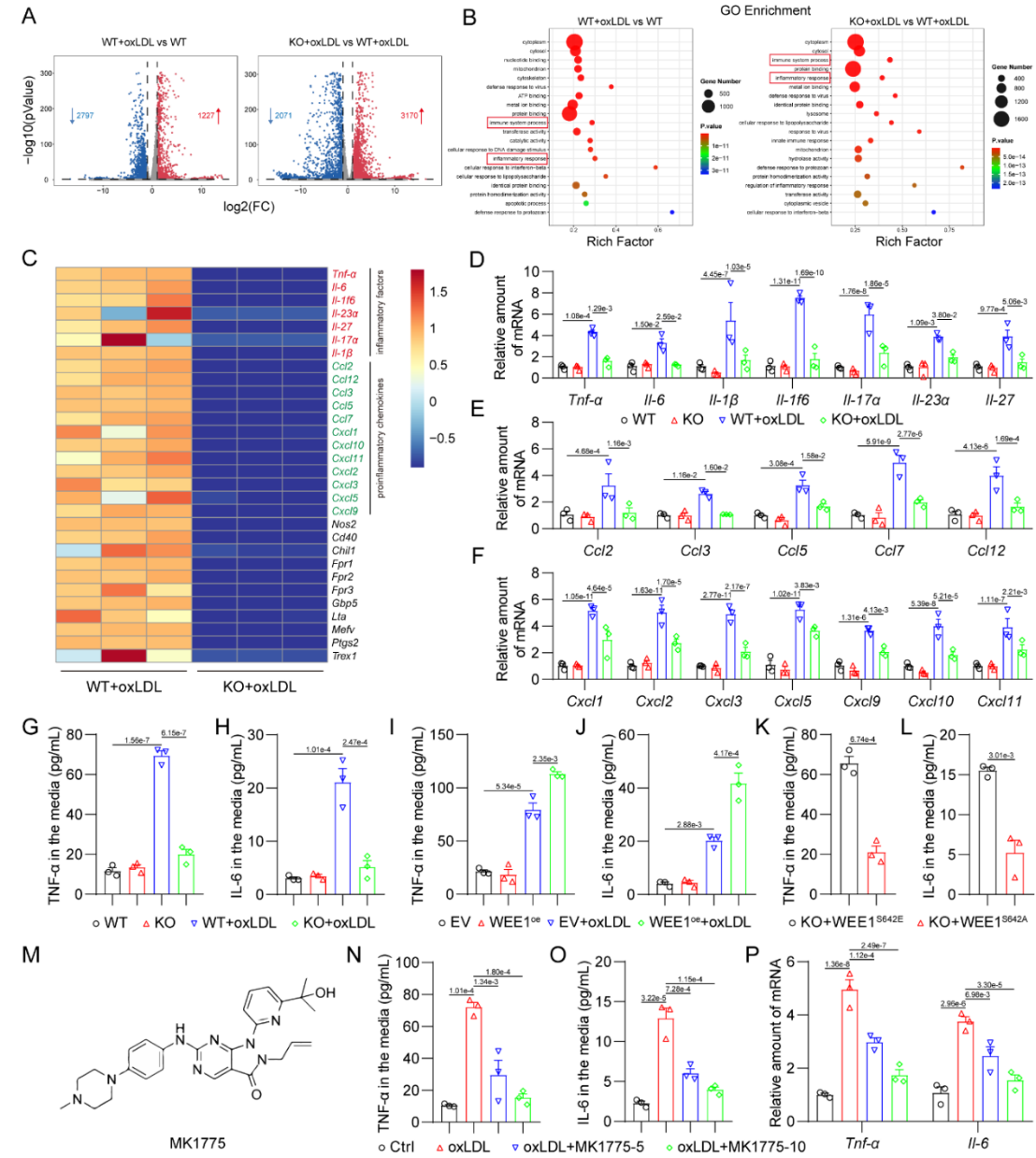
图1. WEE1激活介导了oxLDL诱导的巨噬细胞炎症反应
为进一步探讨巨噬细胞WEE1在动脉粥样硬化中的作用,研究者通过骨髓移植的方式,构建了以ApoE-/-为背景的巨噬细胞特异性WEE1敲除小鼠。HFD喂养小鼠16周后,巨噬细胞特异性WEE1敲除显著减少了HFD喂养诱导的小鼠动脉粥样硬化斑块,并增强了斑块的稳定性(图2)。WEE1的特异性抑制剂MK1775的干预同样可缓解ApoE-/-小鼠动脉粥样硬化进展。
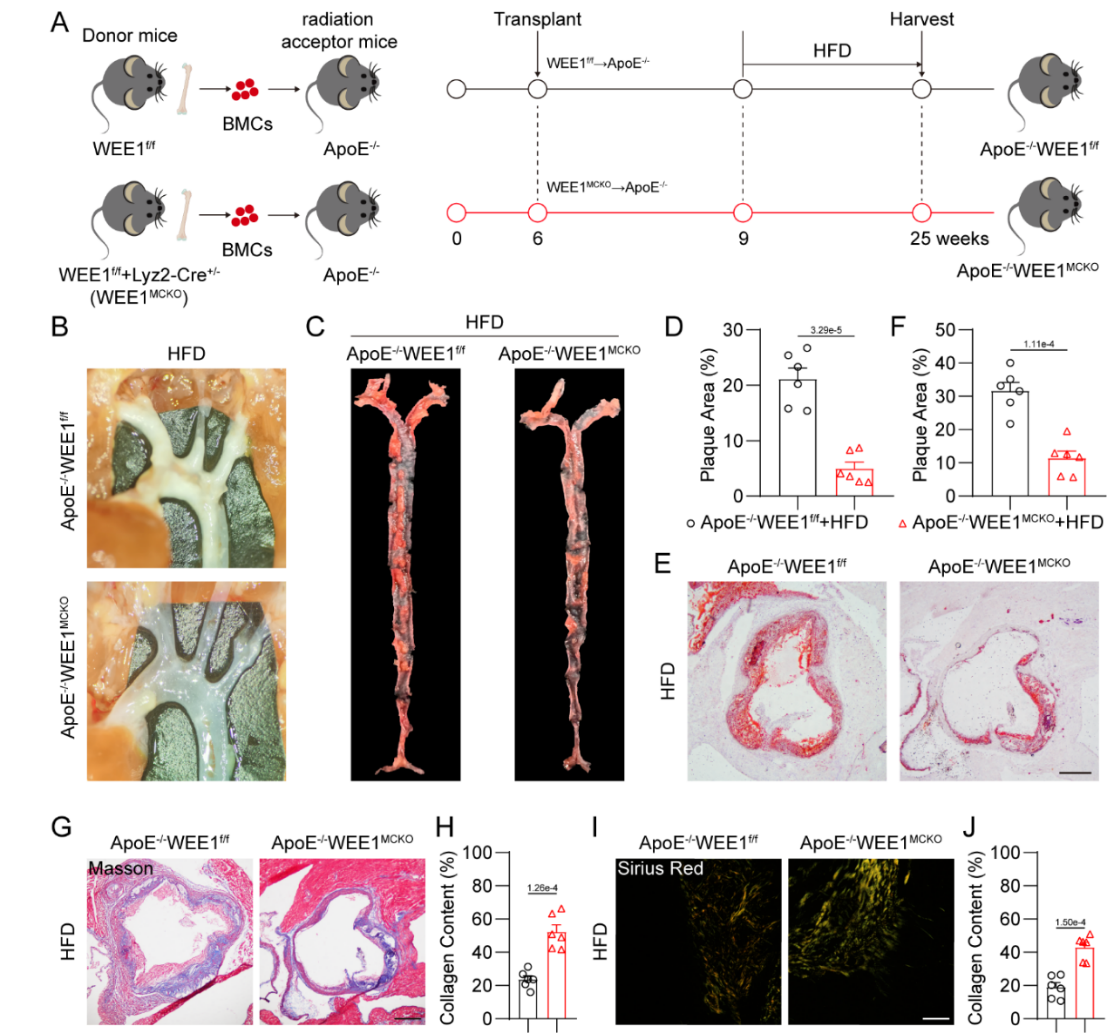
图2. 巨噬细胞特异性WEE1敲除缓解小鼠动脉粥样硬化进展
蛋白激酶通过调控下游通路磷酸化,参与了多种细胞内重要的信号转导过程,与细胞的生物学过程密切相关。为了研究WEE1可能调控的下游信号通路,研究者对RNA测序结果进行富集分析,发现WEE1对NF-κB信号通路具有调节作用。分子生物学实验进一步证实WEE1磷酸化p65的S536位点并激活巨噬细胞NF-κB信号通路及下游炎症因子释放,并且这一调控作用依赖于WEE1的激酶活性。
为了明确WEE1调控p65磷酸化的具体分子机制,研究者通过定量蛋白质组学、免疫共沉淀(Co-IP)、生物膜干涉技术(BLI)、表面等离子共振技术(SPR)等实验筛选并鉴定p65为WEE1的直接结合底物蛋白。研究者进一步确认激活后的WEE1通过激酶结构域与p65的RHD结构域结合,并磷酸化p65的S536位点(图3)。
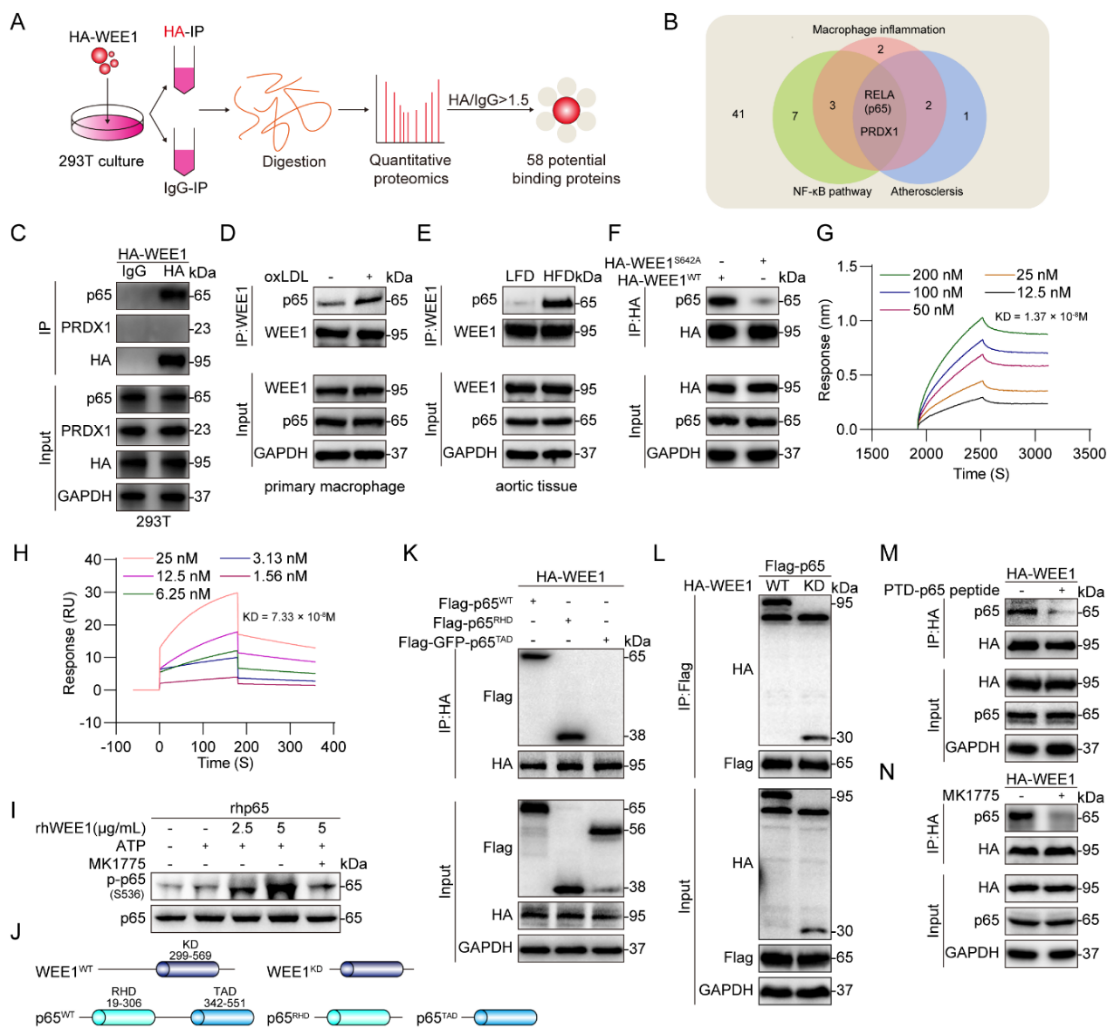
图3. WEE1通过KD结构域与p65的RHD结构域结合,并磷酸化S536位点
综上所述,本研究揭示了蛋白激酶WEE1通过磷酸化p65的S536位点,直接介导巨噬细胞炎症反应及动脉粥样硬化的发生机制,同时揭示了巨噬细胞中直接激活p65的上游激酶和新途径(图4)。这一发现表明WEE1是一个动脉粥样硬化的潜在治疗靶点,针对WEE1的药物抑制或基因治疗有望成为治疗动脉粥样硬化的新策略。
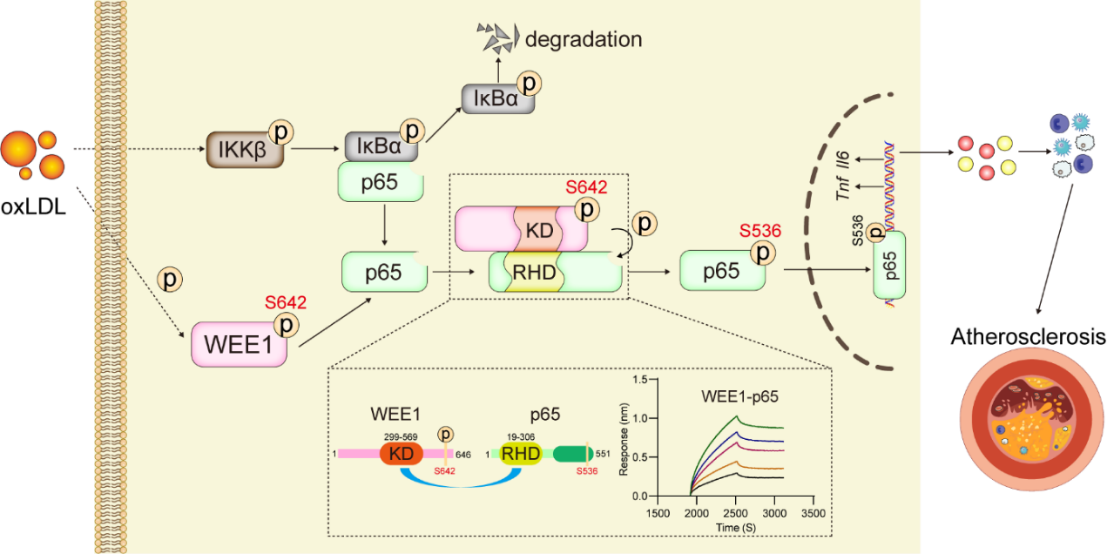
图4. 研究机制图
研究团队在浙江大学、杭州医学院、温州医科大学联合培养的研究生黄祝琦和沈思睿为本论文的共同第一作者。杭州医学院/温州医科大学梁广研究员为本论文末位通讯作者,杭州师范大学王怡研究员为共同通讯作者。
原文链接:
https://advanced.onlinelibrary.wiley.com/doi/10.1002/advs.202503192